Prof. Wenqiang Yu
date:2018-04-19
-
Genome-wide DNA methylation detection and its application in tumorigenesis and metastasis
With the completion of the Human Genome Project, epigenetics has become the frontier of life science research in the post-genome era. DNA methylation is an important part of epigenetics and it plays a crucial role in maintaining normal cell function, embryonic development, disease development, and human tumors. The ENCODE, Roadmap, and HEP(Human Epigenome Program) programs have invested more than 1 billion USD in the identification of apparent markers, but the progress of HEP has been slow, mainly due to the lack of efficient, specific, and cost-effective methods for whole-genome DNA methylation detection. Furthermore, the complete methylation patterns of any certain cell has yet been mapped.
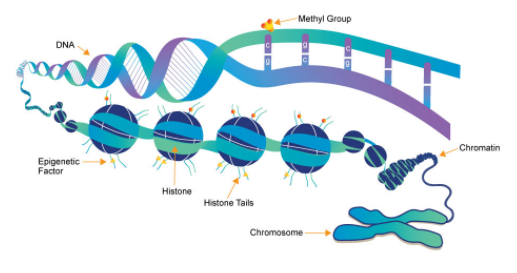
With the support of the National 973 Program, our laboratory developed a genome-wide methylation detection technology named GPS (Guide Positioning Sequencing) with independent intellectual property rights to completely solve the bottleneck of existing methylation detection. This method is capable of: 1) Single-base resolution detection of whole genome DNA methylation sites. 2) Distinguishing between positive and negative strand methylation of DNA templates. 3) Covering almost all replicates of the genome. 4) Significant decrease in testing costs. Using this method, we have successfully established the first complete epigenomic map of human hepatocytes. There is 1.17G cytosine in the human reference genome, and we detected 1.12G cytosines with a coverage rate reaching 96%. Using this technology, we will continue to study the epigenetic changes in the development of high-risk tumors in China, especially the relationship between epigenetic abnormalities, tumor metastasis and drug resistance, and develop single-cell whole genome DNA detection techniques.
-
Screening of tumor epigenetic markers and their applications in liquid biopsy
Thirty years ago, people realized that hypomethylation of whole genome DNA was closely related to the occurrence of tumors. Some time later, people discovered that tumor formation is a contradiction of the hypermethylation of tumor suppressor genes and the genome-wide hypomethylation. However, the exact mechanism of the tumor-specific epigenetic phenomenon has remains unclear. Tumor metastasis is one of the main reasons for the failure of current cancer therapy. Currently, more and more evidence show that epigenetic factors play an important role in tumor metastasis. And though plenty of tumor-associated epigenetic markers have been discovered, few of them are in fact reliable and can be truly applied in clinical diagnosis due to the strong temporal and spatial dynamics of epigenetics. Specifically, when characteristic tumor epigenetics markers encounter inhomogeneity in clinical samples, this generates strong uncertainty and renders the markers useless in actual clinical diagnosis.
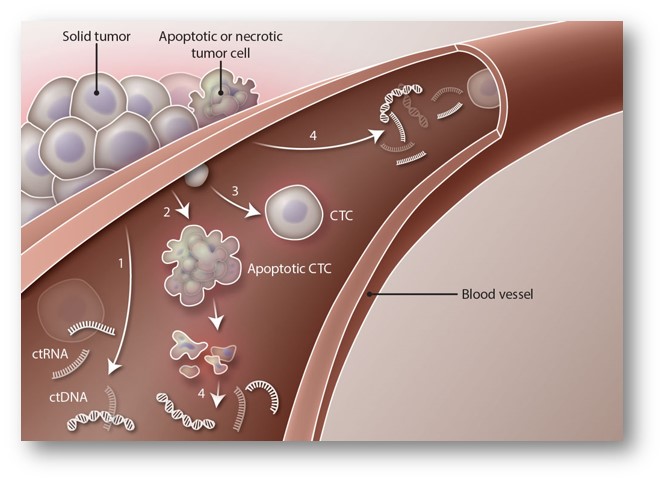
The way of solving this problem relies on finding epigenetic genetic markers common to tumors. For example, DNA methylation is 100% in certain regions of tumor cells and 0% in normal cells. This Yes or No methylation pattern can eliminate the uncertainty in clinical sample detection in clinical diagnosis. At present, we have completed whole genome DNA methylation sequencing of normal human liver tissue, low metastatic liver cancer cell line 97L, and high metastatic liver cancer cell line LM3, identifying a large number of liver cancer-specific DNA methylation abnormalities. Based on this, we compared the methylation data of other tumors and we found that the tumor is characterized by a methylation site, which has been further validated in lung cancer, kidney cancer, prostate cancer, colorectal cancer, bladder cancer, and cervical cancer as well as blood system tumors.
Using this marker, we are able to diagnose tumors through urine, fecal exfoliated cells and cells in cervical smears. We are currently working on carrying out the following research on tumor translational medicine: 1) Select more clinical tumor samples, use GPS to find reliable epigenetic markers that can be widely used in clinical diagnosis. 2) Expand clinical sample size to further validate existing markers, and use tumor liquid biopsy to determine the efficacy and prognosis of cancer therapy. 3) Select patients who have received targeted therapy or immunotherapy and have showed progress, apply our epigenetic methods, and identify reliable epigenetic markers that can predict the effectiveness of clinical therapy in order to truly achieve accurate treatment of tumors.
-
The function of nuclear miRNA and its role in gene activation mediated by enhancers
In 1993, the first miRNA lin-4 was discovered. For 30 years, miRNAs were thought to play a role by down-regulating genes mainly through their 3’UTRs. The function of miRNAs, which are very conservative and have tissue-specific expression, are far from this. 1) At present, the function of the miRNA is at most regarded as ”post-cannon artillery”, always playing a down-regulating effect on the existing genes, and whether the miRNA can regulate gene expression at transcriptional level is an important scientific question. 2) Selective Neglect phenomenon in miRNA research. When miRNA is highly expressed, which is always accompanied by an up-regulation and down-regulation of many genes, people are used to focusing on those down-regulated genes and turn a blind eye to up-regulated genes. If you want to associate miRNAs with up-regulated genes and make sense of it, you must find a repressor, but in most cases, it is difficult to find a suitable repressor. Whether miRNAs can directly participate in gene activation deserves our attention. 3) Missing functional studies of miRNAs in the nucleus.
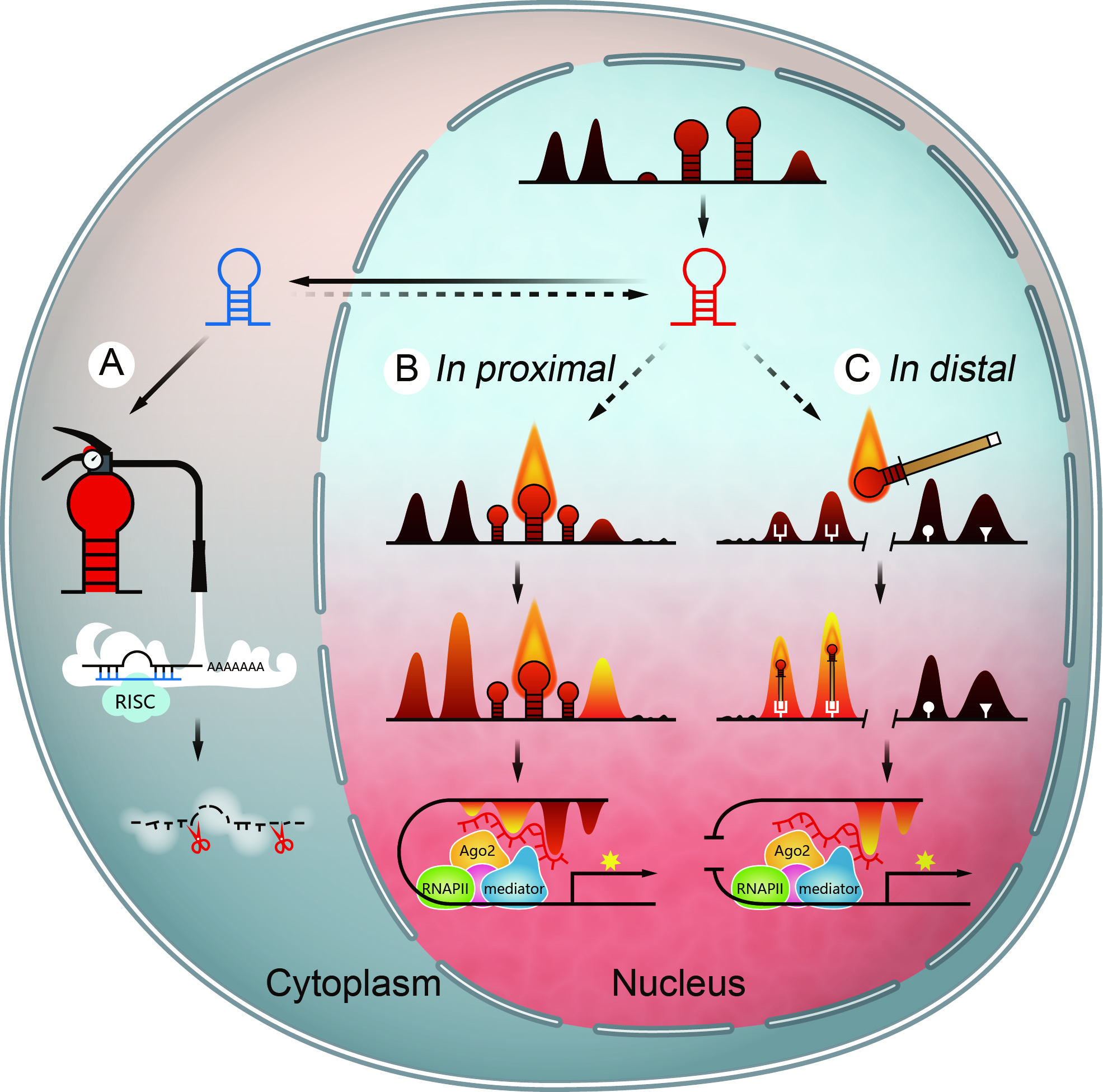
Many evidences indicate that miRNAs can be localized in the nucleus, but how miRNAs play a regulatory role in the nucleus is far from being recognized. Our laboratory study found that a class of miRNAs located in the nucleus can play a positive regulatory role by activating enhancers. We call this the NamiRNA (Nuclear Activating miRNA) which forms the NamiRNA-enhancer-gene activation regulatory network. Different from the traditional miRNA regulation mechanism, it opens up new era for miRNA research and new directions for miRNA and disease research. 1) The molecular mechanism of NamiRNA generation and localization in the nucleus. 2) How the NamiRNA and AGO2 bind to the DNA sequence and thus enhance the activation of enhancers. 3) The role of NamiRNA in the maintenance of cell homeostasis and in the development and metastasis of tumors.